Un cenno di Cinetica Chimica
Le reazioni chimiche conducono alla formazione di nuove sostanze, dette prodotti, a partire da altre sostanze dette reagenti. Il processo avviene attraverso la rottura di vecchi legami e la formazione di nuovi legami; pertanto l'evento reattivo prevede l'incontro tra molecole. A 0°C e 1 atm, la teoria cinetica dei gas prevede che il numero di molecole che si urtano sia pari a circa 1029 molecole/(cm3 sec.); se ogni singolo urto conducesse alla formazione di prodotto, avremmo come risultato che le reazioni avverrebbero tutte con velocità elevatissime. Esistono in realtà alcuni fattori limitanti ed altri discriminanti per le diverse reazioni. Affinché un urto risulti efficace ai fini del processo reattivo è necessario che esso avvenga:
a) con energia sufficiente a provocare la rottura dei legami (energia di attivazione)
b) nella giusta direzione o porzione specifica delle molecole (probabilità sterica)
|
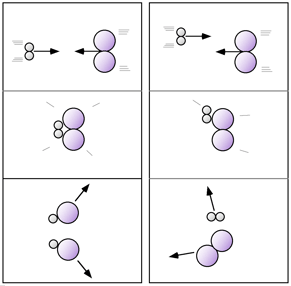
Nella figura a due colonne presente a destra ho cercato di rappresentare, schematicamente, un esempio di urto in direzione efficace (colonna di sinistra) e un esempio di urto non efficace (colonna di destra) per due semplicissime molecole. |
 |
La cinetica chimica è una parte della scienza con carattere squisitamente sperimentale con lo scopo teorico di determinare il meccanismo di una reazione e quello pratico di verificarne l'effettiva fattibilità. Le possibilità di prevedere con buona approssimazione il meccanismo di una reazione sono dettate solamente dall'esperienza di chi lavora da tempo in un campo specifico e con particolari reagenti. La stechiometria di una reazione, infatti, raramente indica il vero meccanismo del processo, che può essere ben più complesso di un pur semplice bilancio stechiometrico. Le reazioni con stechiometria complessa avvengono sempre con una sequenza più o meno numerosa di reazioni elementari.
Quelle reazioni in cui la stechiometria corrisponde con il reale meccanismo sono dette reazioni semplici o elementari. Un esempio viene fornito dalla reazione tra idrogeno e iodio (a temperature non troppo elevate) che avviene con un unico stadio e con il semplice meccanismo di urto bimolecolare come descritto dalla stechiometria del processo:
|
H2 + I 2 |
Come già accennato, il più delle volte le reazioni procedono attraverso un certo numero di eventi semplici (stadi elementari): quello più lento è quello che determina la velocità globale del processo. Le reazioni dell'idrogeno con gli alogeni bromo e cloro, pur avendo medesima stechiometria della reazione precedente, avvengono con un meccanismo radicalico a catena:
|
Br2 Br . + H2 H . + Br2 |
init; |
|
Br . + Br .
H . + H . H . + Br . |
Reazioni di rottura della catena |
Uno dei libri che vi sono stati consigliati (Schiavello) propone il seguente esempio che riguarda una reazione che avviene, alle opportune temperature, in fase gassosa:
|
2 ICl + H2 |
| L'ipotesi che la reazione avvenga in un unico evento, mediante un urto a tre corpi, è altamente improbabile. Infatti, il meccanismo, opportunamente studiato, prevede i due stadi consecutivi appresso riportati: | 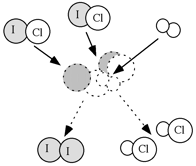 |
|
1° stadio 2° stadio |
ICl + H2 ICl + HI |
Velocità di reazione
Per velocità di reazione si intende la rapidità con cui scompaiono i reagenti o, analogamente, la rapidità con cui si formano i prodotti. La velocità è rappresentata dalla variazione della concentrazione rapportata al tempo in cui si verifica.
|
A + B |
|
Come per i sistemi fisici, la velocità può non essere costante, pertanto è determinata in maniera istantanea come il limite del rapporto Δ[A]/Δt per l'intervallo di tempo tendente a zero, ovvero come la derivata della concentrazione rispetto al tempo. Per un reagente è necessario inserire un segno negativo con lo scopo evidente di rendere positivo il valore della velocità, mentre non è necessario nel caso si consideri un prodotto la cui concentrazione tende a crescere.
|
Per una generica reazione in cui i coefficienti non sono tutti unitari
|
a A + b B |
nel definire la velocità di reazione si deve dividere la derivata per il coefficiente che accompagna la specie, altrimenti osserveremmo velocità diverse per reagenti o prodotti con differente coefficiente stechiometrico (!?).
|
Metodi per misurare la velocità di reazione
Negli studi cinetici è necessario monitorare il valore della concentrazione di una o più specie chimiche durante il procedere della reazione. Molto spesso, invece di determinare il valore effettivo della concentrazione, si utilizza il controllo di una grandezza chimico-fisica ad essa proporzionale ed idonea al tipo di reazione su cui si sta operando.
La tendenza è quella di utilizzare, se possibile, metodi non distruttivi e che non disturbano il corso della reazione stessa. Come indicazione diciamo che i metodi sperimentali più utilizzati riguardano le caratteristiche spettroscopiche (vedi appendice) e potenziometriche delle sostanze ma, ovviamente, nessuna altra possibilità è da escludere.
Ordine e Molecolarità
La velocità di una reazione chimica, misurata istante per istante, dipende da alcuni fattori chimici e fisici. Uno dei fattori importanti è proprio la temperatura; più avanti valuteremo brevemente la sua influenza sulla velocità di un processo chimico.
A temperatura costante la velocità di una reazione tende ad essere una funzione della concentrazione dei reagenti partecipanti al processo chimico. In generale è possibile scrivere una legge di velocità, o equazione di velocità, con una forma simile alla seguente:
| v = k [A] n [B] m |
ove k è la costante di velocità specifica (la velocità riferita a concentrazioni tutte unitarie). Le concentrazioni delle specie interessate al processo risultano elevate ad opportuni coefficienti n ed m, che possono essere diversi dai coefficienti stechiometrici e talune volte sono numeri non interi. Tenendo presente che una legge di velocità è sempre ricavata sperimentalmente, si definisce ORDINE TOTALE (o GLOBALE) di reazione la somma degli esponenti presenti nelle concentrazioni delle sostanze che determinano la velocità del processo (n+m). Come ORDINE PARZIALE si intende l'esponente presente nel termine di concentrazione della singola sostanza presa in considerazione.
NOTA: Il termine ORDINE risulta purtroppo abbastanza abusato, e non solo in chimica. Penso che spesso sia dovuto alla necessità di avere una classificazione sequenziale (es. ordine alfabetico, equazione differanziale di ordine..., cinetica di ordine...., etc), ed in genere per meglio gestire e organizzare certe cose secondo alcune regole e criteri definiti nei vari casi. Sicché si passa dagli ordini professionali alle forze dell'ordine, con l'idea comune di invocare la necessità di un certo tipo organizzazione. La parola "ordine" viene spesso indicata in contrapposizione a "disordine" e questo malauguratamente può provocare cattive interpretazioni di alcune grandezze termodinamiche.
La legge di velocità (e di conseguenza l'ordine di una reazione) viene determinata sperimentalmente e, anche se non pretende di chiarire in maniera assoluta il meccanismo di una reazione, sicuramente può fornire utili indizi a chiarire il modo con cui la reazione si verifica. Noi ci occuperemo solo di processi semplici o sistemi riducibili a quelli semplici.
La MOLECOLARITA' di un processo reattivo, indica il numero di individui chimici che prendono parte all'evento reattivo che determina la velocità globale di un processo. Nelle reazioni che procedono con un meccanismo semplice, ORDINE e MOLECOLARITA' coincidono.
Supponiamo, ad esempio, di avere a che fare con una reazione che procede con un semplice urto bimolecolare (meccanismo corrispondente alla simbologia di scrittura della reazione):
| A + B |
In questo caso la legge di velocità (ricavata sperimentalmente) verrà rappresentata dalle seguente equazione:
| v = k [A] [B] |
Il processo è del secondo ordine globale e la velocità di reazione pertanto dipenderà dalle concentrazione dei reagenti elevate all'esponente unitario. Supponiamo adesso di utilizzare un valore di concentrazione della specie B almeno dieci volte maggiore rispetto a quella della specie A. Alla fine della reazione, mentre la specie A si consuma completamente, la specie B rimane, con una certa approssimazione, quasi invariata, e spesso la sua variazione si dice che rientra nei limiti dell'errore sperimentale. Inglobando la concentrazione di B (costante) nella costante di velocità avremo:
| v = koss [A] |
Quello che osserveremo, monitorando la specie A, è una reazione che procede con un andamento del primo ordine (si dice di pseudo primo ordine, perchè deriva da un processo che globalmente è del secondo ordine).
Il metodo appena descritto può permettere di ridurre l'ordine di una reazione e di isolare l'ordine parziale con riferimento ad uno solo dei reagenti.
Cinetica descrittiva
In questo paragrafo descriveremo il comportamento analitico dei semplici processi corrispondenti a cinetiche di ordine uno, zero e due. Abbiamo accennato come sia possibile che un processo, realmente del secondo ordine, proceda sperimentalmente seguendo il primo ordine anche se la velocità effettiva dipende dalla concentrazione delle due specie. Più enigmatico può sembrare un processo di ordine zero. Come spiegare che un processo non dipende dalla concentrazione del reagente? Ebbene, pur non descrivendo il fenomeno, vi comunico che moltissime reazioni catalizzate dagli enzimi, se effettuate in opportune condizioni, procedono seguendo l'ordine zero, almeno entro certi limiti di concentrazione dei reagenti. Un'altra domanda plausibile è la seguente: Se A e B reagiscono per dare la specie AB e le concentrazioni non sono molto diverse, come è possibile che la reazione proceda seguendo il primo ordine? In questo caso la spiegazione più semplice consiste nell'intervento del solvente che permette un meccanismo alternativo all'urto bimolecolare tra i reagenti indicati nello schema stechiometrico di reazione. Non descriveremo le reazioni con inversa e neppure le reazioni consecutive.
Passiamo ora alle descrizioni analitiche dei processi.
Reazioni del primo ordine
Un processo viene definito del primo ordine quando viene verificata la seguente legge che indica come varia la velocità in funzione della concentrazione:
| v = k [A] |
Per descrivere in dettaglio un processo del primo ordine si confronta la generica espressione della velocità di reazione con quella che si riferisce al processo del primo ordine
|
|||||||
|
Il risultato del confronto fornisce una equazione differenziale del primo ordine che, integrata, permette di determinare come varia la concentrazione della specie chimica interessata (reagente) in funzione del tempo.
|
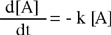
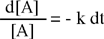
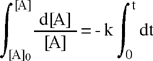
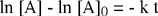
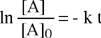
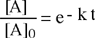
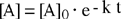
L'integrazione dell'equazione differenziale fornisce come soluzione una funzione di tipo esponenziale:
| [A] = [A]0 e - k t |
Dal punto di vista grafico la velocità di reazione in funzione della concentrazione è rappresentata da una retta passante dall'origine, mentre la concentrazione della specie in funzione del tempo è caratterizzata da un decadimento esponenziale a partire dal valore iniziale di concentrazione, e tende asintoticamente al valore finale.
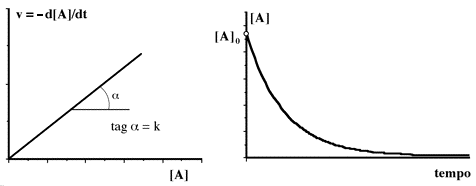
La rapidità con cui la curva si "approssima" al valore finale dipende dalla costante specifica di velocità k (sec-1). Ecco alcuni esempi relativi ad una conc. iniziale 0.1 M con diversi valori di k:
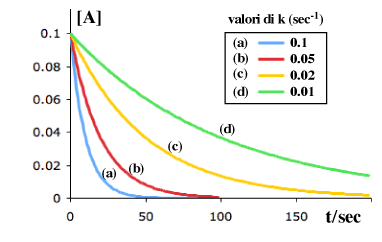
Anche se implicito nella definizione di velocità, ritengo sia utile chiarire il fatto che una reazione del primo ordine non ha "una" velocità ma un numero infinito di valori, da un massimo iniziale ad un minimo pari a zero attraverso tutti i valori intermedi man mano che la concentrazione si riduce.
Periodo di semitrasformazione o tempo di dimezzamento.
Il tempo di dimezzamento è il tempo necessario affinché la concentrazione del reagente considerato dimezzi il suo valore, o, più in generale, affinché il processo in osservazione raggiunga la metà della sua variazione globale a partire dall'istante considerato.
I processi del primo ordine, e la cosa non riguarda solo le reazioni, hanno una particolarità analitica che deriva dalla funzione esponenziale che le caratterizza: il tempo di semitrasformazione è costante per tutta la durata dells reazione ed il suo valore deriva da quello della costante di velocità specifica.
Indichiamo con [A]o/2 la concentrazione dopo un tempo di dimezzamento t2.
|
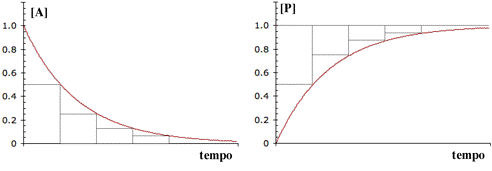
Reazioni di ordine zero
Un processo di ordine zero obbedisce ad una equazione in cui la velocità di reazione risulta indipendente dalla concentrazione:
| v = k [A]0 = k |
Analogamente al caso precedente confrontiamo la generica espressione della velocità di reazione con quella che si riferisce al processo di ordine zero:
|
|||||||
|
Graficamente:
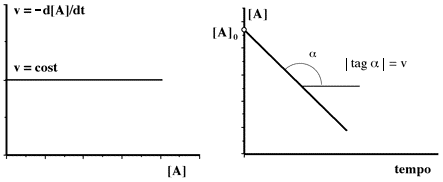
Il primo grafico propone la ovvia indipendenza della velocità dalla concentrazione. Il secondo grafico ripropone la velocità costante in termini di pendenza costante (andamento rettilineo con pendenza negativa). Dal punto di vista analitico la retta dovrebbe toccare, dopo un certo tempo, l'asse delle ascisse; nella realtà si osservano sempre delle distorsioni sia nella parte iniziale che in quella finale. Ad esempio, nelle reazioni enzimatiche, l'andamento di ordine zero è presente solo per concentrazioni elevate di substrato e si osservano alterazioni quando le concentrazioni diventano minori di certe quantità.
Velocità di reazione e temperatura
Come accennato all'inizio di questi cenni di Cinetica Chimica, dai numerosi urti tra le molecole, solo quelli con energia superiore alla "energia di attivazione" possono avere esito favorevole (sempre che l'urto avvenga lungo la direzione giusta). Pertanto solo una parte degli urti sarà efficace ed il processo avverrà gradualmente. Il termine "gradualmente" spazia dai microsecondi ai giorni ed anche oltre. Queste differenze di velocità, talora molto vistose tra differenti reazioni, sono dovute alle diverse energie di attivazione oltre che a dissimili problematiche steriche.
Se non intervengono particolari fenomeni di degradazione termica a carico dei reagenti, dei composti intermedi e di eventuali catalizzatori, tutte le reazioni sono velocizzate da un aumento di temperatura e, viceversa, rallentate da una diminuzione della stessa. Questo comportamento è ovvio se si considera che un aumento della temperatura provoca un aumento dell'energia cinetica media delle molecole, aumentando il numero di urti con energia sufficiente affinché venga superata la barriera di attivazione.
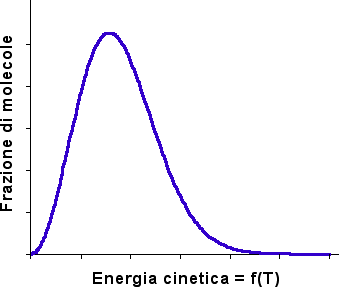 |
Secondo Boltzmann il numero di particelle che statisticamente possiedono energia superiore ad un dato valore (Eatt) viene dato da:
| N(Eatt) = N(tot.) e (- Eatt / R T) |
Il termine esponenziale rappresenta la frazione di molecole aventi energia superiore a Eatt. La costante di velocità di una reazione può essere interpretata con una formulazione similare.
In cui Z (fattore di frequenza) rappresenta il numero di urti, per unità di tempo, che si verificano quando i reattivi hanno concentrazione unitaria. Sebbene questa espressione spiega bene la dipendenza dalla temperatura, fornisce valori troppo elevati e considera "Eatt" come unico fattore discriminante tra due reazioni diverse. Introducendo un altro fattore, detto fattore di probabilità (o fattore sterico) che indica appunto la probabilità che l'urto si verifichi secondo una specifica orientazione spaziale favorevole, abbiamo:
|
Uno studio sistematico della relazione esistente tra la velocità di reazione e la temperatura venne fatto per la prima volta da Svante Arrhenius avvalorando così la teoria dell'urto attivato. Nell'espressione di Arrhenius compaiono come termini esponenziali sia la temperatura che l'energia di attivazione.
| k = A e (- Eatt / R T) |
Il termine pre-esponenziale "A" dipende poco dalla temperatura, ma include il fattore sterico o di probabilità della reazione considerata e il fattore di frequenza degli urti.
TEORIA DELLO STATO DI TRANSIZIONE
Possiamo descrivere una reazione con una particolare simbologia grafica considerando l'energia dei reagenti, quella dei prodotti e la barriera energetica che è necessario superare per "andare" dai reagenti ai prodotti e viceversa (il viceversa riguarda gli equilibri chimici).
La trasformazione che avviene durante un processo elementare passa attraverso uno stato di transizione in cui le specie reagenti formano un "Complesso Attivato", costituito da un'unica molecola (o ione o radicale) per i processi monomolecolari, dall'insieme di due molecole (ioni o radicali) per i processi bimolecolari e così via. La molecolarità quindi rappresenta il numero di particelle reagenti che costituiscono il complesso attivato nello stadio determinante la velocità globale del processo. I processi elementari con molecolarità superiore a 3 in genere non vanno presi in considerazione, perché concorrerebbero scarsamente al verificarsi dell'evento reattivo.
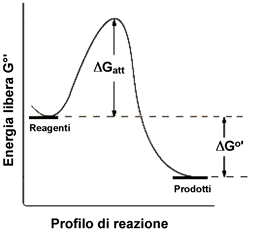
Il complesso attivato è una entità molecolare di vita effimera ma a cui vengono associate sue proprie proprietà termodinamiche. Esso ha una energia potenziale superiore a quella posseduta dai reagenti e dai prodotti. Nella direzione reagenti-> prodotti, la differnza di energia è pari all'energia di attivazione della reazione.
Catalisi e catalizzatori
Alcune sostanze, se inserite nell'ambiente di reazione, hanno la particolarità di accelerare certi tipi di reazione. Queste sostanze si chiamano catalizzatori. Se i catalizzatori hanno la stessa fase dei reagenti si parla di catalisi omogenea, altrimenti si parla di catalisi eterogenea. Nei sistemi biologici esiste un tipo di catalisi estremamente importante (microeterogenea) detta enzimatica perchè sono chiamati "enzimi" quelle sostanze (dal greco ![]() letteralmente "nel lievito") che presentano l'effetto catalitico. Gli enzimi sono macromolecole, in gran parte di natura proteica ma che spesso usano le proprietà dei metalli per attivare sistemi redox.
letteralmente "nel lievito") che presentano l'effetto catalitico. Gli enzimi sono macromolecole, in gran parte di natura proteica ma che spesso usano le proprietà dei metalli per attivare sistemi redox.
| In alcuni testi, per amore di semplicità, viene detto che il catalizzatore agisce fondamentalmente abbassando l'energia di attivazione. Come conseguenza di questo fatto un numero maggiore di urti, a parità di temperatura, avverrà con energia sufficiente a superare la barriera energetica che ora è diminuita. In realtà quello che succede e che la presenza del catalizzatore fornisce una via alternativa alla reazione. I prodotti saranno sempre gli stessi, ma la strada percorsa non è quella che percorre il processo in assenza del catalizzatore. | 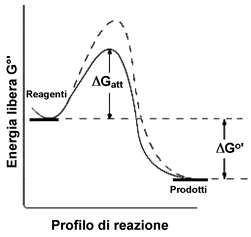 |
Il meccanismo del processo catalizzato è generalmente più complicato e può prevedere un numero maggiore di stadi di reazione, ma tutti con energia di attivazione più bassa rispetto a quella del processo non catalizzato. Si consideri ad esempio la generica reazione:
|
A + B |
ΔGatt elevato; reaz. lenta |
Supponiamo che avvenga con un meccanismo semplice, con un urto bimolecolare tra le sostanze A e B. Se l'energia di attivazione è elevata il processo sarà conseguentemente lento.
Introducendo il giusto catalizzatore nell'ambiente di reazione, la stessa procede anche attraverso il seguente meccanismo alternativo:
|
A + Cat |
ΔGatt basso; reaz. veloce | |
|
A-cat + B |
ΔGatt basso; reaz. veloce | |
|
A + B |
ΔGatt basso; reaz. veloce |
Il catalizzatore, con la sua presenza, facilita in qualche modo la reazione tra A e B, ed alla fine rimane inalterato. Globalmente il processo conduce agli stessi prodotti ma attraverso due stadi di reazione completamente diversi, seguendo un profilo di reazione con due diverse energie di attivazione, entrambe, come già detto, di valore inferiore a quella più elevata prevista per l'urto bimolecolare tra A e B:
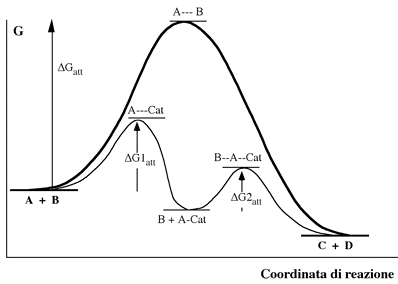
Il meccanismo descritto prevede la formazione di un intermedio di reazione (A-Cat) nel quale interviene direttamente il catalizzatore. Si può pensare che la specie A, quando è legata al catalizzatore si dispone favorevolmente all'attacco della specie B, sia a causa di un allentamento dei legami al suo interno che in termini di probabilità d'urto.
Velocità delle reazioni in soluzione
La presenza del solvente può influenzare in maniera molto marcata la velocità di una reazione, specie se confrontata con un evento reattivo in fase gassosa. In soluzione le molecole dei reagenti sono circondate dalle molecole del solvente che ne ostacolano l'incontro.
Per riuscirci le molecole dei reagenti devono diffondere attraverso il solvente ma, una volta che si incontrano, rimangono ingabbiate e non si allontanano facilmente.
Questo fatto permette un certo numero di urti in posizione diversa aumentando il fattore di probabilità (sterico) a tal punto che alcune reazioni che avvengono in soluzione possono essere veloci quanto e più le corrispettive in fase gassosa.
Le reazioni che avvengono in soluzione e che hanno sia energia di attivazione molto bassa che un fattore sterico unitario (tutti gli urti sono efficaci), sono molto veloci e controllate dalla diffusione: in questi casi è molto importante la viscosità del solvente. La gran parte delle reazioni di trasferimento protonico (acido-base) rientrano in questa categoria.
Se le reazioni avvengono tra specie ioniche, un altro fattore che influenza la velocità del processo è la forza ionica della soluzione. Se gli ioni reagenti hanno lo stesso segno, un aumento della forza ionica aumenta la velocità di reazione; se gli ioni hanno segno diverso, un aumento della forza ionica ne diminuisce la velocità.
APPENDICE
MONITORAGGIO DI UNA REAZIONE MEDIANTE SPETTROSCOPIA UV-VIS
La figura seguente mostra il monitoraggio di una miscela di reazione in soluzione mediante spettroscopia UV-VIS. In generale, dopo un primo approccio spettrale, si registrano solo se variazioni spettrali ad una singola lungezza d'onda scelta tra le più convenienti (es. per avere la più ampia variazione spettrale)
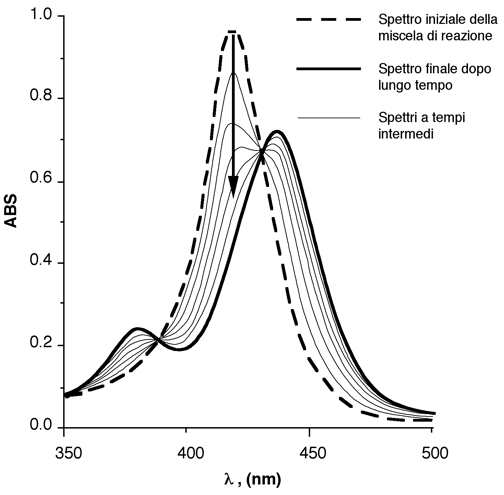
Nella figura seguente viene illustrato un altro esempio in cui si è scelta un specifica lunghezza d'onda per seguire la rapidità del processo cinetico. Nella parte sinistra si vedono gli spettri registrati in tempi successivi; nella parte si sinistra si può osservare l'andamento dell'assorbanza in funzione dello scorrere del tempo
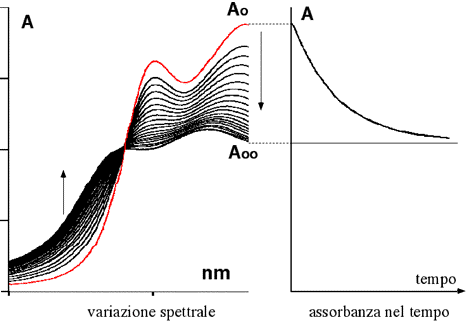
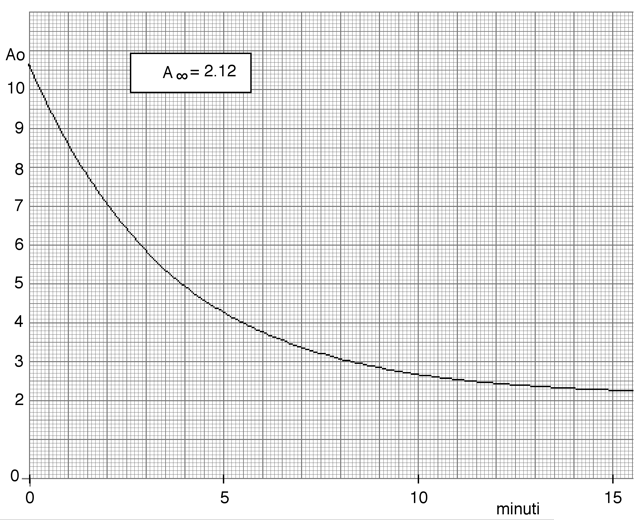
Nella figura seguente viene riportato l'andamento cinetico di una reazione su carta millimetrata. Un buon numero di dati ad essa relativi, sono stati da me estratti e trascritti utilizzando una scala arbitraria delle assorbanze. Verificare che l’andamento è del primo ordine e determinare la costante specifica di velocità.